In areas where chemistry and computer science intersect, a "language" for handling molecular structures on computers is essential. One of the most widely used examples is SMILES notation.
SMILES stands for Simplified Molecular Input Line Entry System. It was proposed by David Weininger in the late 1980s as a notation for representing molecular structures as single-line strings. Today, it is widely used in chemical databases, cheminformatics, AI drug discovery, molecular generation models, and related fields.
For example, ethanol is written as CH3CH2OH as a chemical formula, but in SMILES it can be written as follows.
CCO
This means that carbon, carbon, and oxygen are connected in sequence by single bonds. The strength of SMILES is that it is relatively readable for humans and easy for computers to process. Formats such as MOL files and SDF can store detailed structural information, but when listing large numbers of molecules or using them as input for machine learning models, the compactness of SMILES is a major advantage.
1. Basic SMILES Syntax
SMILES represents molecules by combining atoms, bonds, branches, ring structures, and other elements. The basic symbols are as follows.
| Element | Notation | Example | Meaning |
|---|---|---|---|
| Atom | Element symbol | C, O, N |
Carbon, oxygen, nitrogen |
| Single bond | Usually omitted | CC |
Single bond between carbons |
| Double bond | = |
C=C |
Ethylene |
| Triple bond | # |
C#C |
Acetylene |
| Branch | () |
CC(C)C |
Isobutane |
| Ring structure | Numbers | C1CCCCC1 |
Cyclohexane |
| Aromatic atom | Lowercase | c1ccccc1 |
Benzene |
Atoms commonly used in organic chemistry, such as B, C, N, O, P, S, F, Cl, Br, and I, can be written without square brackets. For these atoms, hydrogen atoms are implicitly added based on standard valence rules.
Examples are shown below.
| Molecule | SMILES | Notes |
|---|---|---|
| Methane | C |
Four hydrogens are implicitly attached to carbon |
| Ethane | CC |
Two carbons connected by a single bond |
| Ethanol | CCO |
Carbon, carbon, and oxygen connected in a straight chain |
| Acetic acid | CC(=O)O |
Contains a carbonyl group and a hydroxy group |
Square brackets are used when you need to explicitly specify charge, isotope, metal atoms, or unusual valence.
- Sodium ion:
[Na+] - Iron(II) ion:
[Fe+2] - Ammonium ion:
[NH4+]
2. Branches, Rings, and Aromaticity
Branches are represented with parentheses. For example, isobutane can be written as follows.
CC(C)C
This indicates that a methyl group branches from the carbon in the main chain.
In ring structures, the same number is placed at two positions to indicate where the ring closes.
C1CCCCC1
This represents cyclohexane. The 1 attached to the first and last carbon indicates that those two atoms are bonded to form a ring.
Benzene can be written in Kekule form as follows.
C1=CC=CC=C1
When aromaticity is expressed explicitly, lowercase c is used.
c1ccccc1
However, aromaticity perception is not completely unique. Software such as RDKit, Open Babel, and Daylight can differ in how they recognize aromaticity and normalize structures. For this reason, when using SMILES in research or data processing, it is important to record the tool and version used.
3. Representing Stereochemistry
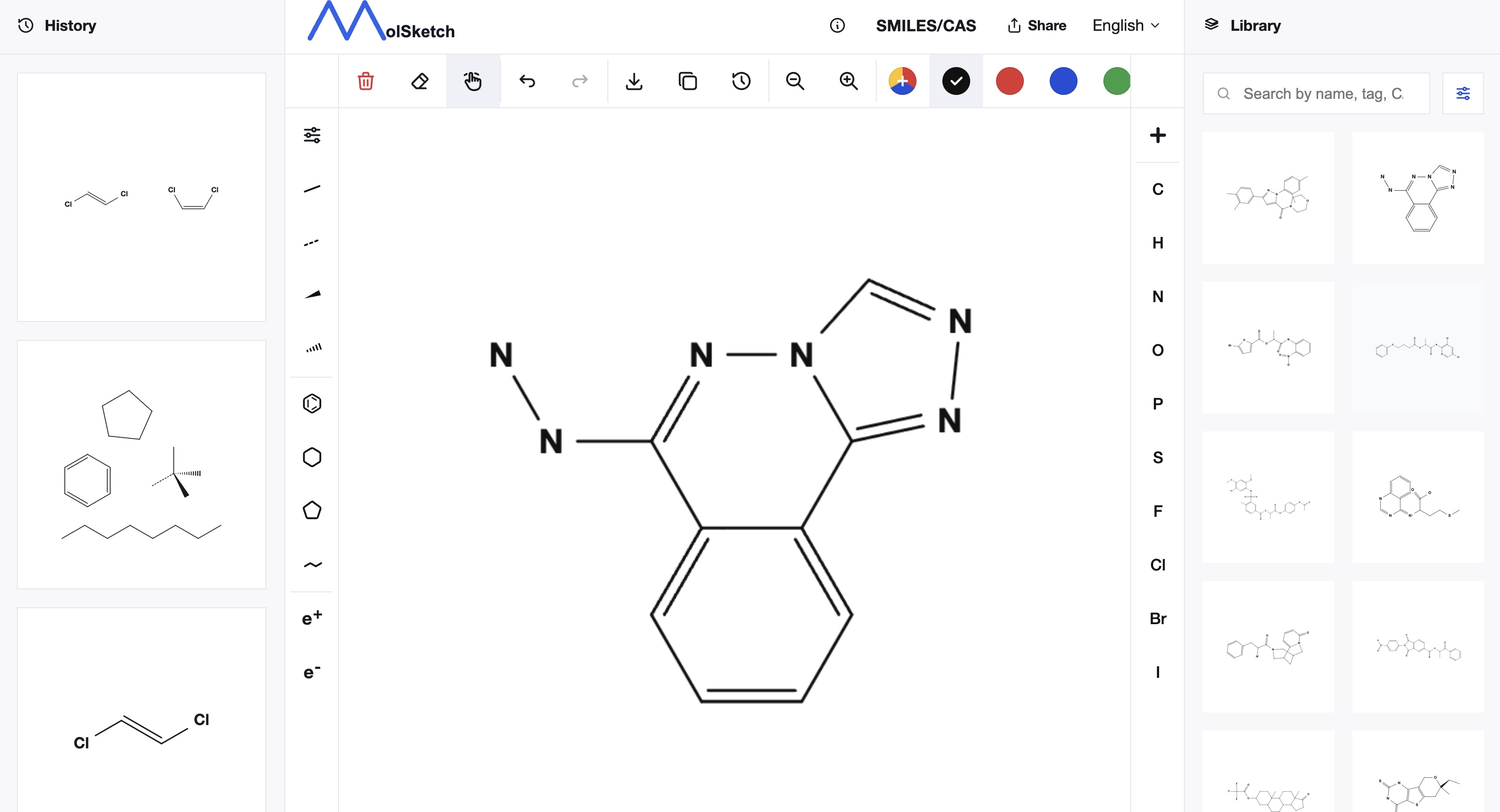
SMILES can represent not only atom connectivity but also some stereochemical information.
| Stereochemical information | Symbol | Meaning |
|---|---|---|
| Chiral center | @, @@ |
Local orientation of a tetrahedral center |
| Geometric isomerism | /, \ |
Relative arrangement around a double bond |
For chiral centers, @ or @@ is used. However, these symbols do not directly mean R or S configuration. They describe local stereochemistry based on the order in which neighboring atoms appear in the SMILES string, so CIP priority rules must be considered separately to determine R/S configuration.
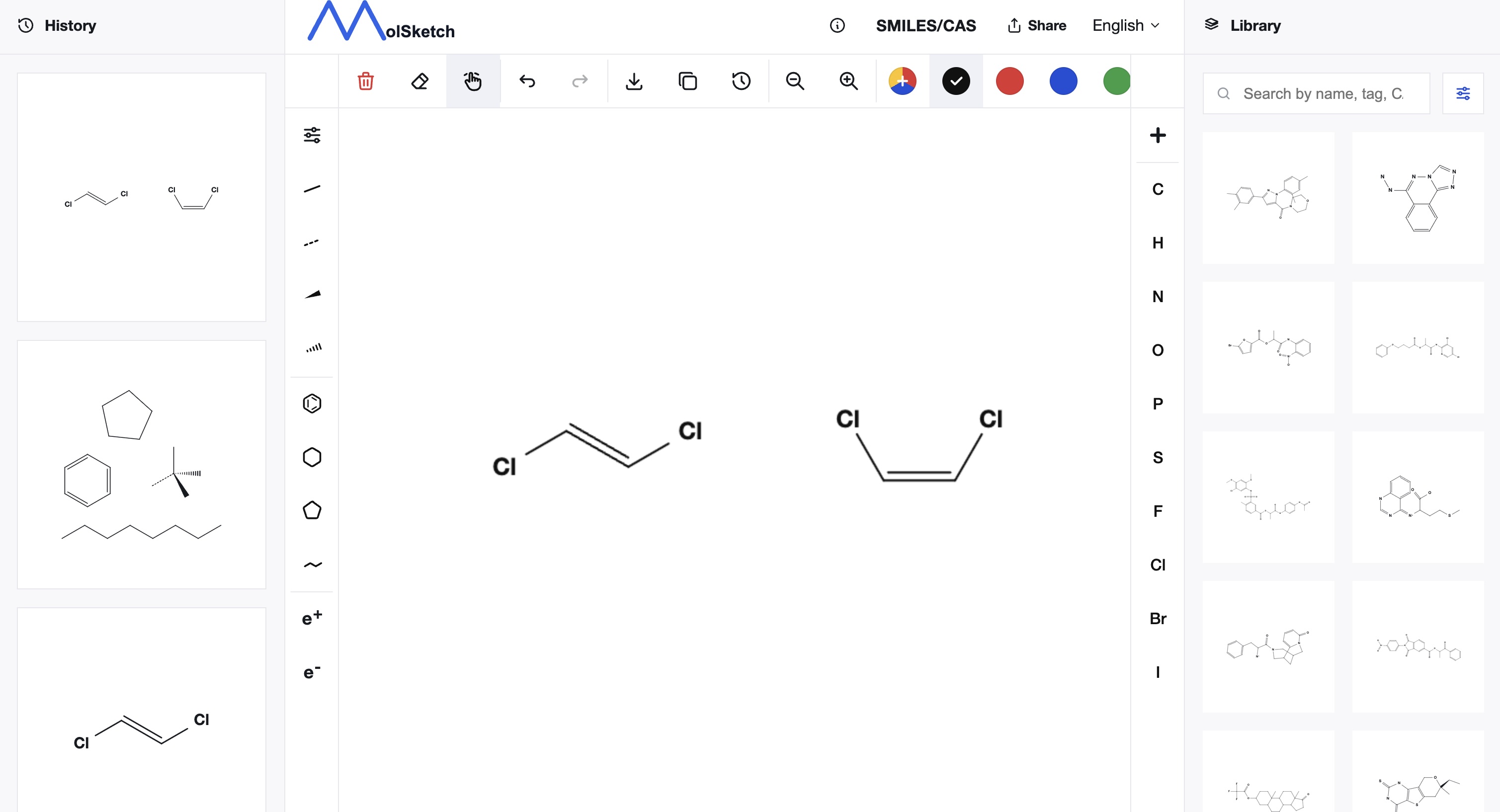
Geometric isomerism around double bonds is represented using / and \.
Cl/C=C/Cl
Cl/C=C\Cl
These notations distinguish isomers of 1,2-dichloroethylene. However, / and \ are also interpreted in the context of the entire string, so their meaning cannot be determined from the symbol alone.
4. Why Canonical SMILES Matters
An important point about SMILES is that the same molecule can have multiple valid representations.
For example, ethanol can be represented in either of the following ways.
CCO
OCC
Both represent the same molecule, but the strings are different. This causes problems in database search and duplicate removal.
This is where Canonical SMILES is used.
| Item | Description |
|---|---|
| Purpose | Make it easier to treat the same molecule as the same string |
| Method | Rank atoms in the molecular graph and generate a representative SMILES |
| Benefit | Useful for duplicate removal, search, and data organization |
| Caution | Output depends on the software implementation |
Canonical SMILES is very useful, but it is not a single fully internationally standardized string. RDKit, Daylight, Open Babel, and other tools may output different normalized SMILES. Therefore, when using it in research, it is best to state which software was used for normalization.
5. Differences from InChI and SELFIES
SMILES is not the only way to represent molecules as strings. Two well-known alternatives are InChI and SELFIES.
| Notation | Main use | Strengths | Cautions |
|---|---|---|---|
| SMILES | Structure description, search, machine learning | Short and relatively readable | Invalid strings can be generated |
| InChI | Compound identification, standardization | Powerful as an identifier | Hard for humans to read |
| SELFIES | Molecular generation, machine learning | Designed so valid molecules are easier to generate | Strings tend to be longer |
InChI (International Chemical Identifier) is a chemical substance identifier developed under the leadership of IUPAC. It describes atoms, bonds, hydrogens, charges, stereochemistry, isotopes, and other information in a layered structure, making it suitable for standard compound identification. On the other hand, it is not suitable for intuitive human reading.
SELFIES (SELF-referencIng Embedded Strings) is a notation proposed with machine learning, especially molecular generation models, in mind. With SMILES, AI systems may generate syntactically invalid strings or chemically inappropriate molecules. SELFIES is designed so that, in principle, any SELFIES string corresponds to a valid molecule.
6. Applications in AI Drug Discovery and Machine Learning
Because SMILES is a string representation, techniques developed in natural language processing can be applied to chemistry. By training models such as RNNs, VAEs, and Transformers on large collections of SMILES, models learn patterns of "chemically plausible strings."
Main applications include the following.
| Application area | Description |
|---|---|
| Molecular generation | Generate new molecular candidates from existing compounds |
| Property prediction | Predict solubility, membrane permeability, toxicity, and other properties |
| Activity prediction | Predict binding to target proteins and pharmacological activity |
| Retrosynthetic analysis | Predict synthetic routes and precursors from a target molecule |
When handling chemical reactions, Reaction SMILES is used. The basic format is as follows.
reactants>agents>products
When the agent field is not used, it is written as follows.
reactants>>products
In retrosynthetic analysis, precursors and reaction routes for synthesizing a target molecule are predicted from the SMILES of that target. This can also be treated as a problem of "translating" a product string into reactant strings.
7. MolSketch Can Automatically Generate Structures from SMILES
In research and development, SMILES is important not only as a "string for storage" but also as an input format for quickly calling up structural formulas.
In MolSketch, entering a SMILES string automatically generates the corresponding chemical structure. If you already have SMILES from a paper, database, internal document, or another tool, you can use it directly as a starting point for creating a structure.
For example, the following workflow is possible.
- Copy a SMILES string you already have
- Paste it into the MolSketch input field
- Automatically generate the structure
- Edit, save, or export it as needed
In this way, MolSketch connects "chemical information as a string" with "chemical structures as diagrams." Instead of drawing everything manually from scratch, it is often more efficient to start from SMILES and adjust only the necessary parts.
It is especially useful for the following cases.
- Quickly turning a compound found in a paper or database into a diagram
- Making small edits based on an existing molecule
- Creating structural formula images for presentations or reports
Understanding SMILES lets you use it not merely as text input, but as an entry point for converting chemical information directly into editable structural formulas.
8. Points to Keep in Mind When Using SMILES
SMILES is convenient, but it is not universal. In particular, the following points require attention.
- Multiple SMILES can exist for the same molecule.
- Canonical SMILES depends on software implementation.
- Aromaticity perception differs between tools.
- If stereochemistry is omitted, isomers may not be distinguishable.
- 3D coordinates and conformational information are usually not included.
- Results can vary depending on how salts, solvents, protonation states, and tautomers are handled.
SMILES is mainly a format for representing molecular graphs and some stereochemical information. When protein binding modes, molecular conformations, solvent effects, and similar 3D information are important, formats such as SDF, MOL2, and PDB, as well as 3D structure generation methods, need to be used together.
Summary
SMILES notation is a concise and powerful chemical language for representing molecular structures as single-line strings. Because it can express atoms, bonds, branches, ring structures, aromaticity, and stereochemistry in short strings, it is widely used in chemical databases, cheminformatics, AI drug discovery, molecular generation models, and other fields.
At the same time, SMILES has the following limitations.
| Limitation | Notes |
|---|---|
| Multiple representations | The same molecule can be represented by multiple strings |
| Implementation dependence | Canonical SMILES and aromaticity perception differ between tools |
| Omitted stereochemistry | Isomers may not be distinguishable |
| Lack of 3D information | Coordinates and conformations are usually not included |
In modern data-driven chemistry, SMILES is a basic interface that allows computers to read and write molecules. Just as chemists draw structural formulas, AI learns molecules through SMILES and explores new candidate compounds. Understanding SMILES will become an increasingly important foundation for researchers involved in chemistry, life sciences, materials science, and AI drug discovery.
References
- Weininger, D. "SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules." Journal of Chemical Information and Computer Sciences, 28(1), 31–36, 1988.
- Weininger, D., Weininger, A., Weininger, J. L. "SMILES. 2. Algorithm for generation of unique SMILES notation." Journal of Chemical Information and Computer Sciences, 29(2), 97–101, 1989.
- Heller, S. R. et al. "InChI, the IUPAC International Chemical Identifier." Journal of Cheminformatics, 7, 23, 2015.
- Krenn, M. et al. "SELFIES: a robust representation of semantically constrained graphs with an example in chemistry." Machine Learning: Science and Technology, 1(4), 045024, 2020.
- Gómez-Bombarelli, R. et al. "Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules." ACS Central Science, 4(2), 268–276, 2018.