In Bereichen, in denen Chemie und Informatik zusammentreffen, ist eine "Sprache" notwendig, um Molekuelstrukturen im Computer zu verarbeiten. Ein typisches Beispiel dafuer ist die SMILES-Notation.
SMILES steht fuer Simplified Molecular Input Line Entry System. Die Notation wurde Ende der 1980er Jahre von David Weininger vorgeschlagen, um Molekuelstrukturen als einzeilige Zeichenketten darzustellen. Heute wird sie in chemischen Datenbanken, der Cheminformatik, der KI-gestuetzten Wirkstoffforschung, molekularen Generierungsmodellen und vielen weiteren Bereichen verwendet.
Ethanol wird als Summenformel zum Beispiel als CH3CH2OH geschrieben, in SMILES jedoch so.
CCO
Das bedeutet, dass Kohlenstoff, Kohlenstoff und Sauerstoff der Reihe nach durch Einfachbindungen verbunden sind. Die Staerke von SMILES liegt darin, dass es fuer Menschen relativ gut lesbar und fuer Computer leicht zu verarbeiten ist. Formate wie MOL oder SDF koennen detaillierte Strukturinformationen speichern, doch wenn viele Molekuele aufgelistet oder als Eingabe fuer Machine-Learning-Modelle verwendet werden, ist die Kuerze von SMILES ein grosser Vorteil.
1. Grundsyntax von SMILES
SMILES stellt Molekuele durch die Kombination von Atomen, Bindungen, Verzweigungen, Ringstrukturen und weiteren Elementen dar. Die wichtigsten Symbole sind die folgenden.
| Element | Schreibweise | Beispiel | Bedeutung |
|---|---|---|---|
| Atom | Elementsymbol | C, O, N |
Kohlenstoff, Sauerstoff, Stickstoff |
| Einfachbindung | Meist ausgelassen | CC |
Einfachbindung zwischen Kohlenstoffen |
| Doppelbindung | = |
C=C |
Ethen |
| Dreifachbindung | # |
C#C |
Acetylen |
| Verzweigung | () |
CC(C)C |
Isobutan |
| Ringstruktur | Zahlen | C1CCCCC1 |
Cyclohexan |
| Aromatisches Atom | Kleinbuchstaben | c1ccccc1 |
Benzol |
In der organischen Chemie haeufige Atome wie B, C, N, O, P, S, F, Cl, Br und I koennen ohne eckige Klammern geschrieben werden. Bei diesen Atomen werden Wasserstoffatome gemaess Standardvalenzen implizit ergaenzt.
Beispiele:
| Molekuel | SMILES | Hinweis |
|---|---|---|
| Methan | C |
Vier Wasserstoffe sind implizit an Kohlenstoff gebunden |
| Ethan | CC |
Zwei Kohlenstoffe mit Einfachbindung |
| Ethanol | CCO |
Kohlenstoff, Kohlenstoff und Sauerstoff in einer linearen Kette |
| Essigsaeure | CC(=O)O |
Enthaelt eine Carbonyl- und eine Hydroxygruppe |
Wenn Ladung, Isotop, Metallatom oder ungewoehnliche Valenz explizit angegeben werden sollen, werden eckige Klammern verwendet.
- Natriumion:
[Na+] - Eisen(II)-Ion:
[Fe+2] - Ammoniumion:
[NH4+]
2. Verzweigungen, Ringe und Aromatizitaet
Verzweigungen werden mit runden Klammern dargestellt. Isobutan kann zum Beispiel so geschrieben werden.
CC(C)C
Dies zeigt, dass eine Methylgruppe vom Kohlenstoff der Hauptkette abzweigt.
Bei Ringstrukturen wird dieselbe Zahl an zwei Stellen gesetzt, um die Ringschlussposition anzugeben.
C1CCCCC1
Das stellt Cyclohexan dar. Die 1 am ersten und letzten Kohlenstoff zeigt, dass diese beiden Atome verbunden sind und einen Ring bilden.
Benzol kann in Kekule-Form so geschrieben werden.
C1=CC=CC=C1
Wenn Aromatizitaet explizit angegeben wird, verwendet man das kleine c.
c1ccccc1
Die Erkennung von Aromatizitaet ist jedoch nicht vollkommen eindeutig. Programme wie RDKit, Open Babel und Daylight koennen sich bei Aromatizitaetserkennung und Normalisierung unterscheiden. Deshalb ist es bei Forschung und Datenverarbeitung wichtig, das verwendete Werkzeug und dessen Version zu dokumentieren.
3. Darstellung von Stereochemie
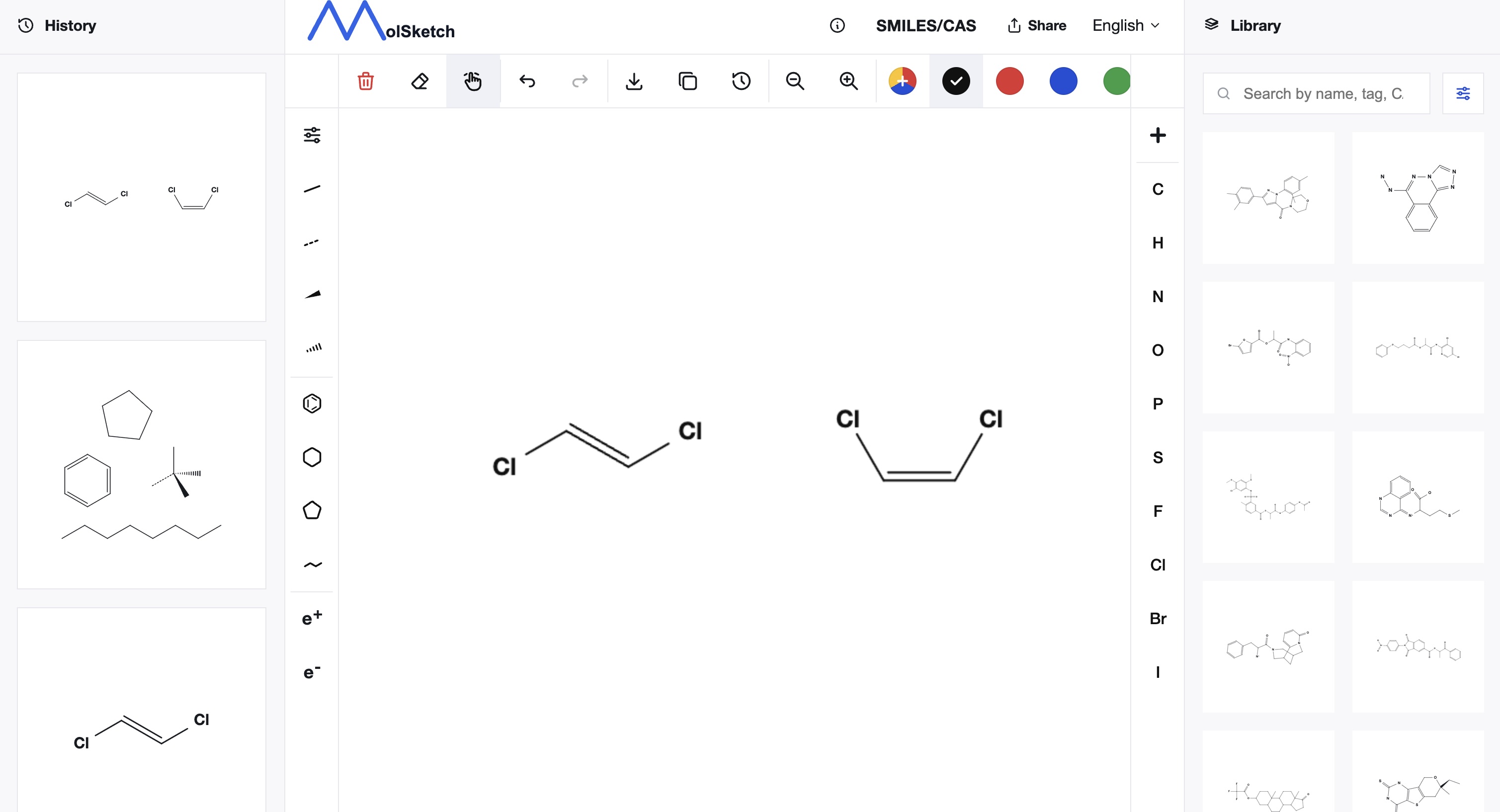
SMILES kann nicht nur Atomverknuepfungen, sondern auch einen Teil der stereochemischen Information darstellen.
| Stereochemische Information | Symbol | Bedeutung |
|---|---|---|
| Chiralitaetszentrum | @, @@ |
Lokale Orientierung eines tetraedrischen Zentrums |
| Geometrische Isomerie | /, \ |
Relative Anordnung um eine Doppelbindung |
Fuer Chiralitaetszentren werden @ oder @@ verwendet. Diese Symbole bedeuten jedoch nicht direkt R- oder S-Konfiguration. Sie beschreiben eine lokale Stereochemie anhand der Reihenfolge, in der Nachbaratome in der SMILES-Zeichenkette erscheinen. Zur Bestimmung von R/S muessen die CIP-Prioritaetsregeln separat beruecksichtigt werden.
Die geometrische Isomerie um Doppelbindungen wird mit / und \ ausgedrueckt.
Cl/C=C/Cl
Cl/C=C\Cl
Diese Schreibweisen unterscheiden Isomere von 1,2-Dichlorethen. Allerdings werden / und \ im Kontext der gesamten Zeichenkette interpretiert, daher kann ihre Bedeutung nicht aus dem Symbol allein bestimmt werden.
4. Bedeutung von Canonical SMILES
Ein wichtiger Punkt bei SMILES ist, dass dasselbe Molekuel mehrere gueltige Schreibweisen haben kann.
Ethanol kann zum Beispiel auf beide Arten dargestellt werden.
CCO
OCC
Beide beschreiben dasselbe Molekuel, sind aber unterschiedliche Zeichenketten. Das verursacht Probleme bei Datenbanksuchen und beim Entfernen von Duplikaten.
Dafuer wird Canonical SMILES verwendet.
| Punkt | Inhalt |
|---|---|
| Zweck | Dasselbe Molekuel leichter als dieselbe Zeichenkette behandeln |
| Methode | Atome im Molekuelgraphen ordnen und ein repraesentatives SMILES erzeugen |
| Vorteil | Nuetzlich fuer Duplikatentfernung, Suche und Datenorganisation |
| Achtung | Die Ausgabe haengt von der Softwareimplementierung ab |
Canonical SMILES ist sehr nuetzlich, aber keine einzige vollstaendig international standardisierte Zeichenkette. RDKit, Daylight, Open Babel und andere Werkzeuge koennen unterschiedliche normalisierte SMILES ausgeben. In Forschungsarbeiten sollte daher angegeben werden, welche Software zur Normalisierung verwendet wurde.
5. Unterschiede zu InChI und SELFIES
SMILES ist nicht die einzige Methode, Molekuele als Zeichenketten darzustellen. Bekannte Alternativen sind InChI und SELFIES.
| Notation | Hauptzweck | Staerken | Hinweise |
|---|---|---|---|
| SMILES | Strukturbeschreibung, Suche, Machine Learning | Kurz und relativ gut lesbar | Ungueltige Zeichenketten koennen entstehen |
| InChI | Identifikation und Standardisierung von Verbindungen | Stark als Identifikator | Fuer Menschen schwer lesbar |
| SELFIES | Molekulare Generierung, Machine Learning | Erleichtert grundsaetzlich die Erzeugung gueltiger Molekuele | Zeichenketten werden oft laenger |
InChI (International Chemical Identifier) ist ein von der IUPAC entwickelter Identifikator fuer chemische Substanzen. Er beschreibt Atome, Bindungen, Wasserstoffe, Ladungen, Stereochemie, Isotope und weitere Informationen in Schichten und eignet sich daher zur standardisierten Identifikation von Verbindungen. Fuer intuitives menschliches Lesen ist er dagegen wenig geeignet.
SELFIES (SELF-referencIng Embedded Strings) wurde mit Blick auf Machine Learning, insbesondere molekulare Generierungsmodelle, vorgeschlagen. Bei SMILES kann KI syntaktisch falsche Zeichenketten oder chemisch ungeeignete Molekuele erzeugen. SELFIES ist so gestaltet, dass prinzipiell jede SELFIES-Zeichenkette einem gueltigen Molekuel entspricht.
6. Anwendungen in KI-Wirkstoffforschung und Machine Learning
Da SMILES eine Zeichenkette ist, lassen sich Methoden aus der Verarbeitung natuerlicher Sprache leicht auf Chemie uebertragen. Wenn Modelle wie RNN, VAE oder Transformer mit grossen Mengen an SMILES trainiert werden, lernen sie Muster "chemisch plausibler Zeichenketten".
Wichtige Anwendungen sind:
| Anwendungsgebiet | Inhalt |
|---|---|
| Molekulare Generierung | Neue Molekuelkandidaten aus bestehenden Verbindungen erzeugen |
| Eigenschaftsvorhersage | Loeslichkeit, Membranpermeabilitaet, Toxizitaet usw. vorhersagen |
| Aktivitaetsvorhersage | Bindung an Zielproteine und pharmakologische Aktivitaet vorhersagen |
| Retrosyntheseanalyse | Synthesewege und Vorstufen aus einem Zielmolekuel vorhersagen |
Fuer chemische Reaktionen wird Reaction SMILES verwendet. Das Grundformat lautet:
reactants>agents>products
Wenn das Agentenfeld nicht verwendet wird, schreibt man:
reactants>>products
In der Retrosyntheseanalyse werden aus dem SMILES eines Zielmolekuels Vorstufen und Reaktionswege fuer dessen Synthese vorhergesagt. Dies kann auch als Problem der "Uebersetzung" einer Produktzeichenkette in Reaktantenzeichenketten betrachtet werden.
7. MolSketch kann Strukturen automatisch aus SMILES erzeugen
In Forschung und Entwicklung ist SMILES nicht nur als "Zeichenkette zur Speicherung" wichtig, sondern auch als Eingabeformat, um Strukturformeln schnell aufzurufen.
In MolSketch kann durch Eingabe eines SMILES automatisch die entsprechende chemische Struktur erzeugt werden. Wenn bereits ein SMILES aus einer Publikation, Datenbank, internen Unterlage oder einem anderen Werkzeug vorliegt, kann es direkt als Ausgangspunkt fuer die Strukturerstellung genutzt werden.
Zum Beispiel ist folgender Ablauf moeglich.
- Vorhandenes SMILES kopieren
- In das Eingabefeld von MolSketch einfuegen
- Die Struktur automatisch erzeugen
- Bei Bedarf bearbeiten, speichern oder exportieren
So verbindet MolSketch "chemische Information als Zeichenkette" mit "chemischer Struktur als Zeichnung". Statt alles von Hand neu zu zeichnen, ist es oft effizienter, mit SMILES zu beginnen und nur die noetigen Stellen anzupassen.
Besonders gut passt das zu folgenden Anwendungsfaellen.
- Eine Verbindung aus Publikation oder Datenbank schnell als Diagramm darstellen
- Ein bestehendes Molekuel als Grundlage nehmen und leicht veraendern
- Strukturformelbilder fuer Praesentationen oder Berichte erstellen
Wer SMILES versteht, nutzt es nicht nur als Texteingabe, sondern als Einstieg, um chemische Information direkt in editierbare Strukturformeln umzuwandeln.
8. Hinweise zur Verwendung von SMILES
SMILES ist praktisch, aber nicht allmaechtig. Besonders folgende Punkte sind zu beachten.
- Dasselbe Molekuel kann mehrere SMILES besitzen.
- Canonical SMILES haengt von der Softwareimplementierung ab.
- Aromatizitaetserkennung unterscheidet sich je nach Werkzeug.
- Wenn Stereochemie weggelassen wird, koennen Isomere ununterscheidbar sein.
- 3D-Koordinaten und Konformationsinformationen sind normalerweise nicht enthalten.
- Ergebnisse koennen sich je nach Behandlung von Salzen, Loesungsmitteln, Protonierungszustaenden und Tautomeren aendern.
SMILES beschreibt vor allem den Molekuelgraphen und einen Teil der Stereochemie. Wenn Proteinbindungsmodi, raeumliche Konformationen, Loesungsmitteleffekte oder andere 3D-Aspekte wichtig sind, muessen Formate wie SDF, MOL2 oder PDB sowie Methoden zur 3D-Strukturgenerierung kombiniert werden.
Zusammenfassung
Die SMILES-Notation ist eine knappe und leistungsfaehige chemische Sprache zur Darstellung von Molekuelstrukturen als einzeilige Zeichenketten. Da sie Atome, Bindungen, Verzweigungen, Ringe, Aromatizitaet und Stereochemie in kurzen Zeichenketten ausdruecken kann, wird sie breit in chemischen Datenbanken, Cheminformatik, KI-Wirkstoffforschung, molekularen Generierungsmodellen und anderen Bereichen genutzt.
Gleichzeitig hat SMILES folgende Grenzen.
| Grenze | Hinweis |
|---|---|
| Mehrere Darstellungen | Dasselbe Molekuel kann durch mehrere Zeichenketten dargestellt werden |
| Implementierungsabhaengigkeit | Canonical SMILES und Aromatizitaet koennen sich je nach Werkzeug unterscheiden |
| Weggelassene Stereochemie | Isomere koennen ununterscheidbar bleiben |
| Fehlende 3D-Information | Koordinaten und Konformationen sind normalerweise nicht enthalten |
In der modernen datengetriebenen Chemie ist SMILES eine grundlegende Schnittstelle, ueber die Computer Molekuele lesen und schreiben. So wie Chemiker Strukturformeln zeichnen, lernt KI Molekuele ueber SMILES und sucht nach neuen Kandidatenverbindungen. Das Verstaendnis von SMILES wird fuer Forschende in Chemie, Lebenswissenschaften, Materialwissenschaften und KI-Wirkstoffforschung immer wichtiger.
Literatur
- Weininger, D. "SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules." Journal of Chemical Information and Computer Sciences, 28(1), 31–36, 1988.
- Weininger, D., Weininger, A., Weininger, J. L. "SMILES. 2. Algorithm for generation of unique SMILES notation." Journal of Chemical Information and Computer Sciences, 29(2), 97–101, 1989.
- Heller, S. R. et al. "InChI, the IUPAC International Chemical Identifier." Journal of Cheminformatics, 7, 23, 2015.
- Krenn, M. et al. "SELFIES: a robust representation of semantically constrained graphs with an example in chemistry." Machine Learning: Science and Technology, 1(4), 045024, 2020.
- Gómez-Bombarelli, R. et al. "Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules." ACS Central Science, 4(2), 268–276, 2018.